
Bez oczu, mózg na wierzchu, serce na zewnątrz. Jak można było kazać kobietom donaszać takie płody?
W debacie o przerywaniu ciąży środowiska anti-choice używają zdjęć uśmiechniętych dzieci z zespołem Downa. To okrutna manipulacja. Pomija straszliwe embriopatologiczne wady płodów: cyklopię, serce na zewnątrz ciała, wynicowanie mózgu
Z okazji 8 urodzin OKO.press, które wystartowało 16 czerwca 2016 roku, przez cały tydzień przypominamy wybrane teksty (i filmy) z tych ośmiu lat. Takie, które zmieniły bieg zdarzeń, ale także po prostu teksty wyjątkowo dobre, ciekawe, ważne dla opinii publicznej. Poza tym mamy dla Was w prezencie osiem wybitnych reportaży OKO.press w wersji audio. I cały czas czekamy na Wasze życzenia, liczymy na wskazanie, czego Wy sobie życzycie od OKO.press
Ten tekst opublikowaliśmy sześć lat temu i szybko stał się jednym z najbardziej czytanych w naszym portalu. A nie jest to łatwa lektura, a przede wszystkim nie jest to łatwy do zniesienia widok. Ludzie kochają takie okropności, powie ktoś. Nie o to raczej tu chodzi – ale o wiedzę, której nikt z nas nie chciałby zdobyć. Potworne wady płodu niestety się zdarzają – i tego nie chcieli nam mówić ci, którzy dążyli do zaostrzenia prawa do aborcji.
Tekst autorstwa Pauliny Łopatniuk, lekarki, specjalistki patomorfologii, która prowadzi popularnonaukowego bloga „Patolodzy na klatce” powstał w odpowiedzi na projekt Kai Godek „Zatrzymać aborcję". Godek chciała, żeby zakazać przerywania ciąży ze względu na ciężkie i nieodwracalne wady płodu. Wtedy ustawa utknęła w Sejmie do końca kadencji. Zmieniła się jednak władza i w październiku 2020 roku Trybunał Konstytucyjny Julii Przyłębskiej uznał, że przesłanka embriopatologiczna nie jest powodem do przerwania ciąży.
Oznaczało to, że kobiety będą musiały donaszać i rodzić zdeformowane płody – takie, o jakich pisze Paulina Łopatniuk. Zwycięzcy wyborów 15 października obiecywali zmianę prawa, ale różnice między koalicjantami (Lewica i Koalicja Obywatelska chcą dopuszczenia aborcji do 12 tygodnia, Trzecia Droga powrotu do tzw. kompromisu) powodują, że stan prawny pozostaje de facto bez zmian (w praktyce można usunąć ciążę ze względu na stan psychiczny kobiety, która dowiaduje się o takiej wadzie).
Dlatego ten tekst sprzed sześciu lat wciąż jest boląco aktualny.
Uwaga! Artykuł ilustrowany jest drastycznymi zdjęciami!
Przeczytaj także:
Bezocze, mózg poza czaszką, zarośnięcie przełyku. „Brzydkie dzieci” z wadami wrodzonymi, które trzeba będzie rodzić
Posługiwanie się zdjęciami uśmiechniętych dzieci z zespołem Downa to żadna nowość. Pamiętamy to chociażby z 2016 roku, kiedy to Bogdan Chazan, wówczas dyrektor szpitala, odmówił rodzicom aborcji. Do jego placówki miała pecha trafić ciężarna pragnąca skorzystać z ustawowej możliwości terminacji ciąży ze względu na poważne i nieuleczalne wady płodu.
Nieszczęsną matkę z przymusu oskarżano o chęć „zabicia dziecka za to, że brzydkie”.
Zresztą „argumenty”, że podłe „aborcjonistki” po prostu wstydzą się wychowywać chore dzieci co jakiś czas przewijają się przez dyskusję.
To przecież okropne, prawda? Taki „urodyzm”, zwłaszcza że przecież możemy te biedne, brzydkie płody leczyć, żeby stały się odrobinę ładniejsze, prawda?
Nie powinnam kpić, to sprawa poważna. Śmiertelnie wręcz poważna i wiążąca się z prawdziwymi tragediami, czego chyba wolą nie zauważać zwolennicy i zwolenniczki zaostrzenia naszej restrykcyjnej ustawy. Inaczej nikt przecież nie pozwalałby sobie na drwiące z wiedzy medycznej uwagi o leczeniu wielu wad, o których mowa. Zwłaszcza gdy mówimy o tych uznawanych za letalne, a to najbardziej dramatyczny wątek dyskusji.
Bolesną kpiną jest mówienie o aborcji z przesłanek embriopatologicznych jako o aborcji eugenicznej. Eugenika w wersji opracowanej przez Francisa Galtona miała służyć odgórnej, sterowanej przez władze, poprawie materiału genetycznego – rasy, narodu, gatunku, społeczności. Przypisywanie podobnych intencji kobietom albo rodzicom podejmującym decyzję o przerwaniu ciąży po rozpoznaniu ciężkiej nieuleczalnej choroby u płodu, jest nie tylko błędne, nie tylko głupie, ale też – jak to niedawno słusznie podsumował w wywiadzie dla Gazety Prawnej genetyk, profesor Lucjusz Jakubowski – zwyczajnie podłe.
Skoro jednak nie o brzydotę chodzi i nie o uśmiech na radosnej twarzyczce o nieco szczególnych rysach kojarzonych z nadmiarowym chromosomem 21, to o co właściwie chodzi w całych tych protestach, w krzyku wokół wad letalnych, w zamieszaniu wokół dziecka, które przyszło na świat dzięki rozrostowi sumienia pana Chazana?
Co jeszcze dolega płodom, na które zamach tak oburza środowiska przeciwne prawu wyboru?

Multum wad
Nie każda wada rozwojowa jest w Polsce – choćby i teoretycznie, bo z realizacją ustawowych wyjątków bywa różnie – podstawą do przerwania ciąży. Definicja wrodzonej wady rozwojowej jest szeroka i pojemna. Nikt nie próbuje terminować ciąży z powodu palca dodatkowego czy wyrośli przedusznej, nieprawidłowości dosyć częstych.
Mianem wad letalnych, najistotniejszych w debacie, zwykło się określać te najcięższe zaburzenia rozwojowe, których rokowania są złe. I nie chodzi o rokowania dotyczące leczenia, tylko przeżycia. Te zaburzenia zwykle prowadzą do zgonu wewnątrzmacicznego, czyli – zależnie od etapu ciąży – do poronienia lub urodzenia martwego dziecka. A nawet w przypadku żywego urodzenia zazwyczaj powodują zgon w okresie niemowlęcym, bardzo rzadko tylko pozwalając przeżyć dłużej, jednak z dramatycznymi niekiedy obciążeniami.
Doskonałym przykładem jest tu niedaleki krewny zespołu Downa – zespół Edwardsa: 95 proc. płodów nie dotrwa narodzin. Połowa tych, które się urodziły, nie przeżyje tygodnia. Do końca okresu niemowlęcego dobrnie jakieś 10 proc. Co setne dożyje dziesiątego roku życia. Pojedyncze dożyją dorosłości. Bardzo szczególnej dorosłości.
Dlaczego piszę o niedalekim krewnym zespołu Downa? Bo to ta sama grupa wad. Letalne trisomie obejmują chromosomy autosomalne, czyli inne niż chromosomy płciowe (X i Y). Taki typ zaburzeń obejmujący nie zmiany pojedynczych genów, a całego zestawu chromosomów (choć mniejsze niż pomnożenie całego ich garnituru) nazywamy aneuploidią.
Trisomia jest szczególną postacią aneuploidii. U zdrowego człowieka każda komórka poza gametami, czyli komórkami rozrodczymi, zawiera podwójny zestaw chromosomów. Dwie sztuki każdego spośród 22 niezależnych od płci chromosomów autosomalnych i jedna para chromosomów płciowych w każdej komórce ciała. Trisomia oznacza, że któregoś z tych 23 jest o jeden więcej.
Czyli trzy chromosomy 21 w zespole Downa. Trzy chromosomy 18 w zespole Edwardsa. Trzy trzynastki w zespole Pataua. Te trzy zespoły najczęściej spotyka się u żywo urodzonych dzieci, bo możliwości jest oczywiście tyle, co chromosomów. Trisomię 21 jako najmniej spośród nich destrukcyjną spotykamy najczęściej, goni ją zespół Edwardsa i nieco rzadszy zespół Pataua.
Inne porodu doczekują niezwykle rzadko – w materiale poronieniowym obok trisomii 21 królują potrojone chromosomy 16 i 22. Poważne malformacje narządów wewnętrznych uniemożliwiają im dłuższe funkcjonowanie nawet w tych nielicznych przypadkach, gdy dziecko przeżyje poród. Wady serca i przewodu pokarmowego, brak lub ciężki niedorozwój nerek, zniekształcenia mózgu.
Bez oczu, z jednym okiem, z wieloma palcami, ze zrośniętym przełykiem
Spójrzmy jednak na te, które przeżywają częściej.
W zespole Edwardsa dominującą cechą u tych dzieci, które przeżyją pierwsze chwile, będzie głęboka niepełnosprawność intelektualna.
Towarzyszyć jej będą poważne wady serca (u dziewięciorga na dziesięcioro dzieci), napady drgawkowe i zaburzenia kostno-szkieletowe. Atrezja (czyli zarośnięcie) przełyku w wielu przypadkach uniemożliwi normalne karmienie bez dodatkowych – niekiedy poważnych – zabiegów.
Zaburzenia dróg oddechowych mogą utrudnić postępowanie anestezjologiczne, a pamiętajmy, że opieka paliatywna będzie tu podstawą. Ostatecznie najprawdopodobniej śmierć przyjdzie pod postacią niewydolności oddechowej.
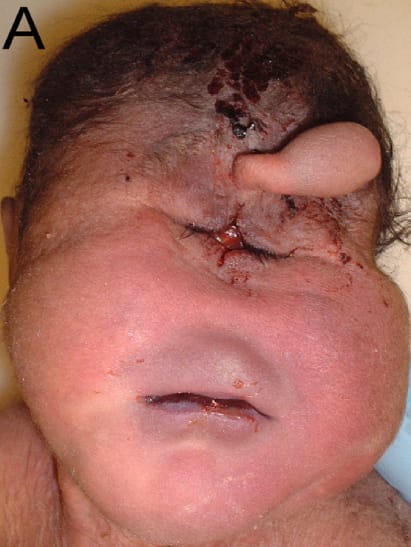
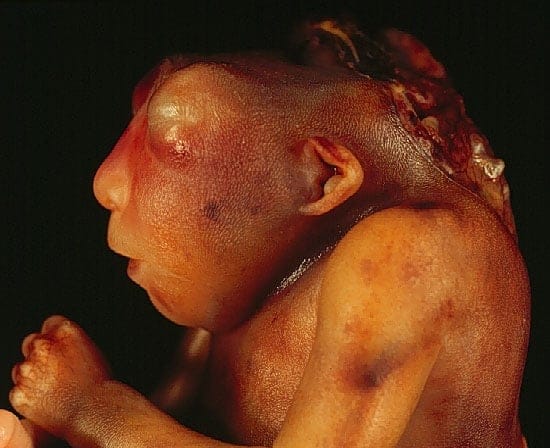
Obecność dodatkowego chromosomu 13 obok niepełnosprawności intelektualnej obejmuje holoprozencefalię z wadami budowy twarzy i towarzyszącymi wadami narządu wzroku o szerokim spektrum – od bezocza (anoftalmia) po cyklopię. Pomijając klasyczną dla wady triadę małogłowia, polidaktylii (wielopalczastości) i rozszczepów podniebienia.
Holoprozencefalia, czyli przodomózgowie jednokomorowe (całkowity lub częściowy brak podziału mózgu na półkule), to z kolei nie tylko olbrzymia niepełnosprawność intelektualna, ale też cały bukiet upośledzeń psychomotorycznych i deficytów neurologicznych. Napady drgawkowe w różnym stopniu odpowiadające na leczenie farmakologiczne. Zaburzenia napięcia mięśniowego. Zaburzenia endokrynologiczne wynikające z nieprawidłowej budowy przysadki mózgowej i podwzgórza.

„Bąbelki”
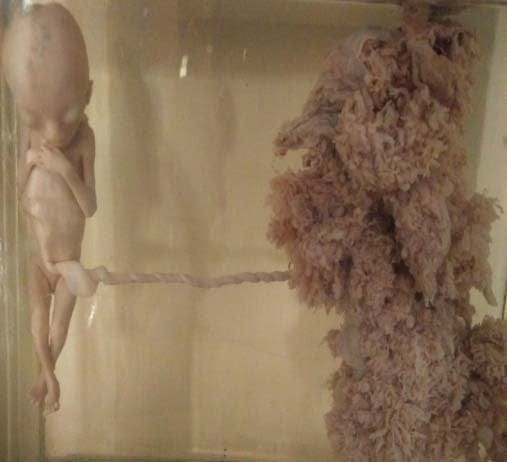
Skoro jeden nadmiarowy chromosom potrafi narobić takiego bałaganu, co było, gdyby pomnożyć cały ich zestaw? Gdyby zamiast podwójnego pakietu 23 chromosomów, zarodkowi przydarzył się zestaw potrójny? W sytuacjach tego typu zaburzeń mówimy o poliploidii. Nie jest to zjawisko niespotykane w przyrodzie: poliploidna pszenica jest standardem na naszych polach, w ogóle zresztą rośliny są dość tolerancyjne, jeśli chodzi o liczbę chromosomów i poliploidyzacja nie jest wśród nich rzadka, przy czym nie mówię tylko o tych uprawnych.
Ze zwierzętami jest gorzej, choć nadal istnieją wyjątki, a pośród ryb w ogóle nie jest to rzadkość. Ale człowiek nie jest ani rybą, ani pszenicą i znosi podobne sytuacje fatalnie. Najładniejszym przykładem są tu zaśniady groniaste. Nie te całkowite, te akurat, słynne bąbelki, nie mają przed sobą żadnych perspektyw pomimo liczbowo prawidłowego zestawu chromosomów. Szczęśliwie większość nawet zdeterminowanych przeciwników przerywania ciąży, dzieci w nich nie widzi, nawet brzydkich.
W zaśniadzie częściowym w większości przypadków dwa plemniki zapładniają zwykłe zdrowe jajeczko, dając początek nieco mniej niźli w zaśniadzie całkowitym wątłemu człowiekowi o potrójnym zestawie chromosomów i ten niestety do pewnego stopnia się rozwinie. Ciężarna nie będzie miała jednak okazji zastanowić się, czy, „wstyd go będzie komuś pokazać”, bo zazwyczaj poroni, a w tych nielicznych przypadkach, gdy ciąża zakończy się żywym urodzeniem dziecko przeżyje nie więcej niż parę miesięcy.
Oczywiście nie trzeba zaraz uszkodzeń, braków czy nadmiarów całych chromosomów, by ciąża nagle okazała się katastrofą. Istnieje całe mnóstwo letalnych wad jednogenowych. Ot, taka chociażby dysplazja tanatoforyczna, zwana nie bez powodu dysplazją śmiertelną. Jedna z najczęściej występujących letalnych dysplazji kostno-szkieletowych.
Ciężki zespół wad wrodzonych spowodowany mutacją w genie FGFR3 kodującym receptor czynnika wzrostu fibroblastów-3. To kluczowe dla rozwoju kości białko reguluje wzrost kości i kostnienie chrząstek, co pozwala częściowo przynajmniej przewidzieć konsekwencje jego uszkodzeń. Przedwcześnie zarośnięte szwy czaszkowe, zwężona hipoplastyczna klatka piersiowa, skrócenie kości kończyn długich. Skoro niedorozwinięta jest klatka piersiowa, to i narządy w niej zawarte, czyli płuca chociażby. Skoro mamy do czynienia ze zmianami w czaszce, to i zaburzenia rozwoju układu nerwowego nie powinny dziwić. I rzeczywiście – zwężenie otworu potylicznego wielkiego z niewydolnością ośrodka oddechowego zlokalizowanego w uciśniętym rdzeniu przedłużonym w połączeniu z niedorozwojem płuc to jedna z głównych przyczyn zgonu w przypadkach, które nie zakończyły się zgonem wewnątrzmacicznym.
Tak, w pojedynczych przypadkach dzieci te dożywają dorosłości. Dorosłości wymagającej wspomagania oddechu, dorosłości obciążonej ograniczeniami motorycznymi, licznymi chorobami współistniejącymi i rozwojem intelektualnym na poziomie dziecka kilku-, może kilkunastomiesięcznego.
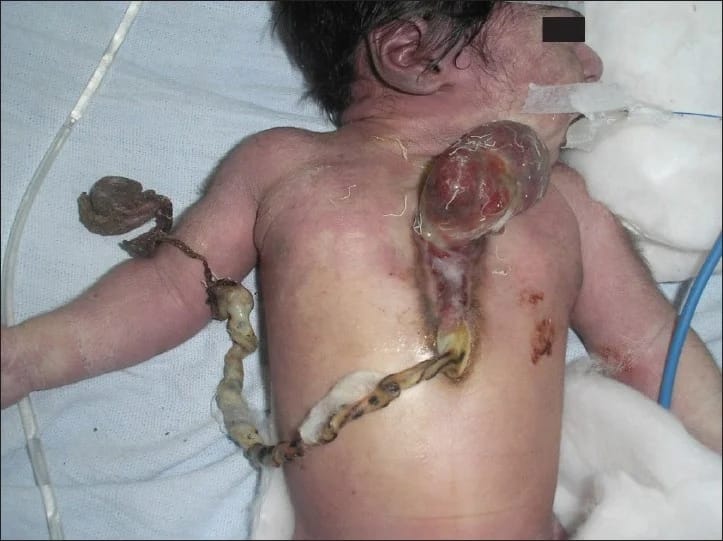
Serce na zewnątrz ciała
A niekiedy nie ma żadnych konkretnych genów, których uszkodzenia można by wychwycić, żadnych łatwych do obarczenia winą zaburzeń genetycznych, a jednak efekty nieprawidłowości w rozwoju płodowym trudno zlekceważyć. Tak będzie w niektórych zaburzeniach rozwoju serca. Nie, nie mam na myśli prostych wad typu defektów przegrody międzykomorowej czy międzyprzedsionkowej. Mówię o zaburzeniach z kręgu ektopii, gdy cały narząd znajduje się poza wnętrzem klatki piersiowej, zwisając na szypule naczyniowej na przykład przez dziurę w mostku. Ktoś się oburzy i powie, że rzecz przecież się leczy. I rzeczywiście, zależnie typu wady i od położenia takiego wyrwanego ze swej zwykłej lokalizacji narządu osiąga się pewne sukcesy.
W zeszłym roku na przykład doczekano się pierwszego w Wielkiej Brytanii dziecka (urodzona w listopadzie 2017 Vanellope Hope Wilkins), które udało się utrzymać przy życiu mimo obciążenia taką wadą, a za najstarszą żyjąca osobę urodzoną z ektopią serca uchodzi urodzony w 1975] Amerykanin, Christopher Wall –
w ciągu pierwszych osiemnastu miesięcy życia poddano go piętnastu zabiegom chirurgicznym mającym na celu przemieszczenie jego serca w bardziej adekwatną lokalizację.
Nie ma oczywiście mowy o przywróceniu dziecku pełnej sprawności – ani samo serce zwykle nie jest w pełni prawidłowe, ani rozwijające się bez niego wnętrze klatki piersiowej nie przewiduje dlań miejsca. No i nadal to przypadki nieliczne. Większość takich historii kończy się zgonem niedługo po narodzeniu, a personelowi medycznemu i rodzinie pozostaje skupienie się na zapewnieniu noworodkom możliwie godnych warunków umierania, na opracowaniu odpowiedniego opatrunku, by nagi pozbawiony nawet pokrywy skórnej narząd zwyczajnie nie wysechł, czy na odpowiednim zaopatrzeniu przeciwbólowym.
I nie znamy konkretnych czynników przyczyniających się do rozwoju ektopii serca, nie mamy też twardych papierów na jakieś konkretne zaburzenia genetyczne. Wiemy, że pewne zaburzenia chromosomalne mogą ektopii serca sprzyjać, owszem, ale niewiele więcej.
Wbrew nadziejom polityków czy aktywistek chcących wady letalne leczyć nie mamy tu żadnego punktu zaczepienia, by zadziałać w pierwszych tygodniach rozwoju zarodkowego, gdy wada zaczyna się kształtować.

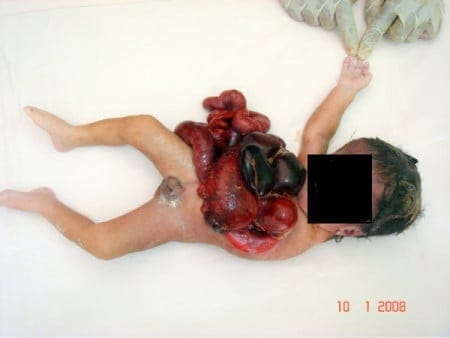
Bez ściany brzucha, bez czaszki. Mózg poza czaszką
Nie jesteśmy też pewni przyczyn powstawania zmian w niewinnie brzmiącym zespole braku pępowiny (LBWC, limb-body wall complex). Co drastycznego w braku pępowiny? Ot, skrót myślowy. Nie tylko pępowina jest tu bowiem problemem, a właściwie wcale nie ona. W klasycznym obrazie zespołu brakuje nie tyle pępowiny, ile ściany brzucha w ogóle, a defekt sięga niejednokrotnie i klatki piersiowej – ektopia serca będzie tu co najwyżej drobnym dodatkiem do obrazu całości wady, wisienką na torcie.
Wnętrznościom znajdującym się poza ciałem, niejednokrotnie pozrastanym z powierzchnią łożyska, towarzyszą zaburzenia rozwoju szkieletu osiowego i kończyn. W części przypadków dochodzą do tego wady rozwoju cewy nerwowej. To wady układu nerwowego związane z tworzeniem się i zamykaniem pierwotnej cewy nerwowej, tego zawiązka, z którego później rozwiną się mózg i rdzeń kręgowy – od prostych przepuklin oponowych, oponowo-rdzeniowych czy mózgowych po bezczaszkowie (akrania), wynicowanie mózgu (łac. exencephalia) z mózgowiem obecnym (choć zwykle poważnie uszkodzonym) w worku oponowym lub skórnym poza czaszką i bezmózgowie (łac. anencephalia).
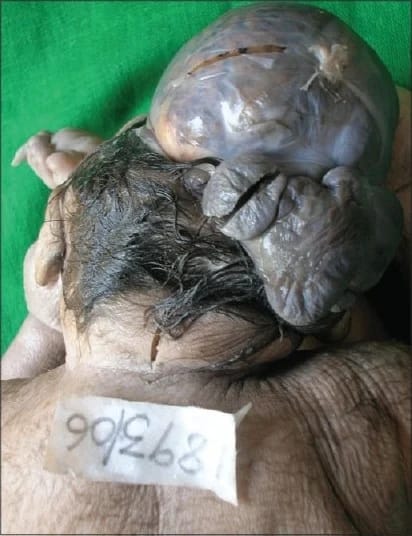
Bez mózgu
Do wad rozwoju cewy nerwowej, czyli dysrafii, nie potrzeba od razu całego zespołu braku pępowiny ani żadnych dużych wad chromosomalnych. Powstają one także jako wady izolowane. Sporej części dysrafii jesteśmy w stanie zabiec, pamiętając o suplementacji kwasu foliowego u ciężarnych i unikaniu pewnych leków.
Nie wszystkim jednak. Niektóre można ze znakomitymi efektami leczyć operacyjnie,
bezmózgowia czy wynicowania mózgu jednak nie wyleczy się, bo i jak.
Bezmózgowie jest zresztą przykładem wady bardzo obecnej w narracji okołoaborcyjnej. Strona postulująca pozbawienie kobiet prawa wyboru chętnie operuje tymi kilkoma przypadkami dzieci z bezmózgowiem, które przeżyły dłużej niż kilka dni czy tygodni.
Krążą zdjęcia maluchów o uszkodzonych czaszkach przykrytych dzierganymi czapeczkami mające zadać kłam ciężkości zaburzenia. Samo hasło „bezmózgowie” zresztą też może być nieco mylące dla laika zastanawiającego się nad możliwością życia bez mózgu. Mimo braku półkul mózgowych czy móżdżku pozostają dziecku przynajmniej szczątkowe struktury tych najprymitywniejszych części mózgu odpowiedzialnych za oddychanie chociażby. Taki maluch potrafi czasami przy dużej dozie szczęścia czy pecha – zależnie od tego, jak na to spojrzeć – trwać nieco dłużej. A że bez jakiejkolwiek nawet perspektywy i w oczekiwaniu na nieunikniony zgon, to już inna rzecz.
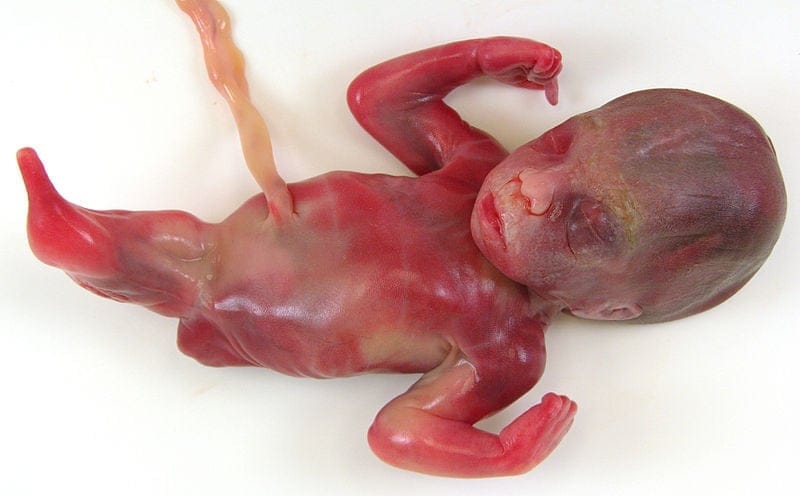
Bez nóg
Mała syrenka kojarząca się baśniowo niekoniecznie wzbudzi podobne skojarzenia genetyków czy neonatologów. Syrenomelia zwana zespołem syreny lub syrenowatością kończyn również zazwyczaj kończy się zgonem, czy to wewnątrzmacicznym, czy następującym krótko po urodzeniu. I nie do końca wiadomo, skąd się bierze, choć w części przynajmniej przypadków podejrzewa się nieprawidłowości naczyniowe. U zdrowego zarodka, rozwijają się dwa pączki czy zawiązki, z których następnie formują się kończyny dolne, w syrenomelii jest inaczej. Choć zlanie się zawiązków kończyn w docelowo jedną przypominającą syreni ogon może się wydawać wadą przykrą ze względu na ograniczenia ruchowe, nie śmiertelną jednak, wrażenie to jest złudne.
Zazwyczaj do syreniego ogona dochodzą poważne deformacje miednicy i jej narządów, z agenezją (niewykształceniem) nerek, niedorozwojem pęcherza moczowego czy przewodu pokarmowego (ot, zarośniecie odbytu chociażby czy niedorozwój jelita w ogóle), z kloaką dającą wspólne ujście przewodowi pokarmowemu, drogom moczowym i pochwie.
Tak, niektóre z tych zaburzeń da się czasem naprawić chirurgicznie. W łagodniejszych postaciach wady kończyny dolnej współtworzące ogon bywają tak wysoce rozwinięte, że można je nawet rozdzielić (w cięższych nie ma czego rozdzielać, bo mamy do czynienia z pojedynczą kością udową), w innych kolejne operacje pozwalają wytworzyć nowy pęcherz moczowy i wyłonić odbyt, dając choremu dziecku szanse na życie zbliżone do normalnego.
Ale nie ma co się łudzić – to przypadki wyjątkowe i literatura notuje kilka zaledwie przypadków dłuższego przeżycia małych syrenek.
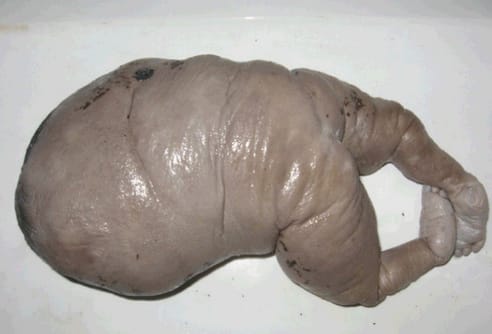
Bez serca
Zaburzenia naczyniowe to też podstawa jednej z najbardziej chyba spektakularnych, poza zroślakami, patologii ciąży bliźniaczej. Wspólne dla obu płodów łożysko oznacza ryzyko zaistnienia połączeń naczyniowych (anastomoz) częściowo łączących krążenia pępowinowe bliźniąt, połączeń zarówno powierzchownych, bezpośrednich, pomiędzy poszczególnymi żyłami i tętnicami, jak i pośrednich, głębokich, zachodzących na poziomie naczyń włosowatych.
Anastomozy te mogą przyczyniać się niekiedy do nieprawidłowego, nieproporcjonalnego przepływu krwi do poszczególnych płodów, co z kolei może owocować całym wachlarzem patologii, w tym zespołu odwróconej perfuzji tętniczej, TRAP (twin reversed arterial perfusion). To, że jeden z płodów rozwija się bez głowy nie jest tu wbrew pozorom największym problemem, gorszy jest brak serca (nie, nie metaforyczny wcale). Krążenie płodu bez serca w obliczu tego podstawowego defektu napędzane jest zasadniczo dzięki sercu zdrowego bliźniaka (zwanego z tego właśnie względu) bliźniakiem pompującym, pump twin. Pasożytując na krwiobiegu drugiego bliźnięcia, osłabia je i stwarza ryzyko jego obumarcia. Nieleczony zespół odwróconej perfuzji tętniczej oznacza zgon pompującego bliźnięcia w mniej więcej połowie przypadków. O ryzyku zdrowotnym dla bliźniaka bez serca nie ma co pisać – on nie ma szans na przeżycie, ma tylko szanse pociągnąć za sobą brata lub siostrę.
Nie zawsze oczywiście, jednak szanse zdrowego bliźnięcia powiększa odcięcie odeń pasożytniczego rodzeństwa, przerwanie nieprawidłowego przepływu krwi pomiędzy bliźniętami w drodze koagulacji. Skutkiem bezpośrednim jest uśmiercenie bezsercowca, dyskusyjne zapewne w obliczu postulowanych zmian w ustawie, które wszak miałyby dopuszczać jedynie ratowanie życia ciężarnej.
Ogrom cierpienia
Nie ma sensu ciągnąć tej wyliczanki, wadom wrodzonym poświęcono całe podręczniki, których przepisywanie byłoby żmudne dla przepisujących i nużące dla zmuszonych do czytania. Rzecz w tym, że większość poważnych wad wrodzonych to nie kwestia urody czy brzydoty tudzież wstydu przed pokazaniem ludziom.
To ogrom cierpienia towarzyszącego zarówno obojgu rodzicom świadomym przyszłych losów oczekiwanego dziecka, jak i samej ciężarnej skazanej na donoszenie skazanego na śmierć płodu. To również niemożliwa do oszacowania perspektywa samego dziecka, które umrze zaraz po urodzeniu bądź może nawet i przeżyje dni, miesiące czy lata skazane na mechaniczną wentylację i ciągłą osłonę przeciwbólową, a w przypadkach nieco „lepiej” rokujących na dziesiątki operacji i tak kończących się poważnym upośledzeniem.
Nawet „oswojony” już przecież i niezaliczany do wad letalnych zespół Downa, to życie trudne zarówno dla dziecka, jak i jego opiekunów, zwłaszcza gdy towarzyszą mu częste w trisomii 21 wady serca czy przewodu pokarmowego. Cóż dopiero rzec o życiu półtorarocznego dziecka uwięzionego w ciele niezdolnego do oddychania bez wspomagania mechanicznego dwudziestoparolatka.
Jedyne co akceptowalne w takiej sytuacji, to wsparcie osób, przed którymi stoi trudna decyzja o dalszym postępowaniu po usłyszeniu diagnozy. Wesprzeć zarówno w decyzji o ewentualnej terminacji, jak i w zapewnieniu wszelkiej możliwej pomocy w razie postanowienia donoszenia ciąży. Tu jest miejsce na opiekę psychologiczną i medyczną, na hospicja perinatalne, na zabezpieczenie paliatywne, na dostęp do niezbędnego sprzętu medycznego, etc.
Ale najpierw musi być wybór, bez presji i bez opowieści o dzieciobójstwie. Bo wzruszająca opowieść o maleństwie, które „ma rurkę tracheostomijną, jak nasz papież” i które z miłością „patrzy swoim jednym oczkiem” pokazuje tylko jedną stronę historii.
Nie każdego rodzica stać psychicznie na zachwyt tym, że dziecko, które przyszło na świat „miało wszystko – przełyk, podniebienie, całą główkę”, a potem walkę z dziesiątkami dla większości z nas niewyobrażalnych problemów zdrowotnych i cierpliwe oczekiwanie na śmierć niemowlęcia, mimo że, jak to zasugerował jeden z polityków, wszyscy przecież umrzemy.
Komentarze